
- Academic Editors
Alzheimer’s disease (AD), a primary cause of dementia, is rapidly emerging as one of the most financially taxing, lethal, and burdensome diseases of the 21st century. Increasing evidence suggests that microglia-mediated neuroinflammation plays a key role in both the initiation and progression of AD. Recently, emerging evidence has demonstrated mitochondrial dysfunction, particular in microglia where precedes neuroinflammation in AD. Multiple signaling pathways are implicated in this process and pharmaceutical interventions are potentially involved in AD treatment. In this review, advance over the last five years in the signaling pathways and pharmaceutical interventions are summarized and it is proposed that targeting the signaling pathways in microglia with mitochondrial dysfunction could represent a novel direction for AD treatment.
Alzheimer’s disease (AD) is an ambient neurodegenerative disorder distinguished
by memory deficits and diverse mental impairments. It manifests in a variety of
cellular alterations such as dysregulation of microRNAs, activation of glial
cells and astrocytes, hormonal imbalances, impaired mitophagy and synaptic
deterioration, as well as the accumulation of intracellular neurofibrillary
tangles (NFTs), extracellular
AD is characterized as a dual-proteinopathy disorder, exhibiting a spatially
selective distribution of diffuse neuritic A
The etiology of AD continues to be a
controversial topic and is not fully understood [10, 11, 12]. Current models suggest
that the accumulation of neurotoxic A
Microglia, as the resident macrophages of the central nervous system (CNS), are
crucial for maintaining CNS homeostasis. They have been implicated in AD
pathology and participate in phagocytosis, removal of pathogenic molecules and
modulate neuroinflammatory responses [25, 26, 27, 28, 29]. Activation of microglia is a
critical element in the pathogenesis of AD [30]. Robust experimental data suggest
a role for activated microglia in A
Mitochondria play an important role in oxidative stress, cell survival, calcium homeostasis and energy metabolism [35]. Mitochondrial dysfunction is considered to be directly involved in the death of autonomic neurons [35]. AD has been thought to be a metabolic disease [28, 35, 36, 37]. Although glial mitochondria are more metabolically flexible than neurons, they also play a critical role in the function of the CNS and have specific metabolic and mitochondrial characteristics to support their functional diversity [28, 35, 38]. Several studies have identified a range of mitochondrial dysfunctions in microglia and other brain cells, including age-related accumulation of mitochondrial DNA (mtDNA) mutations, structural remodeling, overproduction of mitochondrial reactive oxygen species (ROS), changed mitocondrial membrane potential, reduced ATP levels, disruptions in the electron transport chain, and elevated mitochondrial fragmentation, resulting in defective mitophagy [1, 31, 39, 40, 41, 42, 43, 44]. These mitochondrial dysfunctions contribute to the onset of AD. Further research has revealed drugs targeting various signaling pathways related to mitochondrial energy metabolism, dynamics, biogenesis, and autophagy.
This review outlines recent advancements in understanding the relationship between microglial mitochondrial dysfunction and AD. It also summarizes the signaling pathways involved and pharmaceutical interventions targeting these pathways over the past five years. Lastly, we introduce a novel perspective for targeting specific signaling pathways as a therapeutic approach for AD.
Serving as the resident macrophages of the CNS, microglia maintain CNS homeostasis; they are also implicated in AD pathology [25, 45]. Upon exposure to pathogenic or damage signals, microglia rapidly activate to acquire different phenotypes exerting either neuroprotection or neurotoxicity [46]. As brain resident innate immune cells - microglia - sculpt neural circuitry and coordinate copious and numerous neurodevelopmental requirements [47].
Microglia, acting as effector cells in the brain’s innate immune system, exhibit
an adaptability to perform various specialized functions in AD, including
phagocytosis and elimination of harmful clusters of A
Microglia express glycolysis and oxidation and in the absence of glucose, they are metabolically flexible and have the capacity to quickly adapt to consume glutamine as an alternative metabolic fuel. Glutamine hydrolysis supports the maintenance of motor and damage sensing functions of microglia processes in insulin-induced hypoglycemia in blood glucose or in vivo in acute brain sections. This metabolic adaptation sustains mitochondrial function and is regulated through mTOR-dependent signaling. This plasticity allows microglia to maintain their critical monitoring and phagocytosis roles even after disruption of neural energy homeostasis [52, 53]. Glucose deprivation may sensitize microglia to release inflammatory mediators and major microglia functions in survival and inflammation, which probably result in psychiatric comorbidities of ischemia, diabetes, and/or metabolic disorders [54]. Microglia also undergo a transition from a resting phenotype to a reactive phenotype, known as disease-associated microglia. There is a common misconception that M1, the glycolytic type, and M2 are dependent on the oxidative phosphorylation phenotype. The glucose uptake of microglia is mainly promoted by glucose transporter type 1 (GLUT1), especially within inflammatory environments [55].
Microglia continuously monitor the brain parenchyma to detect changes in neuronal activity and homeostatic processes to maintain normal brain function. Metabolic pathways participating in microglial activity adapt and promote cell phenotypes. While mitochondrial oxidative phosphorylation is efficient in the production of ATP, microglia produce ATP faster and produce intermediates for cytokine production and cell growth. In macrophages, pro-inflammatory stimulation induces a metabolic transition from oxidative phosphorylation to glycolysis, a phenomenon similar to the Warburg effect in tumor cells. Alterations in metabolic function allow macrophages to respond appropriately to changing environments and much evidence suggests that, similar to macrophages, microglia are able to variably adapt to the energy matrix. Neuroinflammation frequently manifests in various neurodegenerative disorders and metabolic reprogramming of microglia has been found in neurodegenerative diseases [56].
Mitochondria serve as the primary energy-generating organelles in human cells. The genetic blueprint for mitochondria, mitochondrial DNA (mtDNA) is a circular, double-stranded molecule comprising 16,569 nucleotides. It encodes 13 messenger RNAs, 2 ribosomal RNAs and 22 transfer RNAs [57].
Proteins transcribed from mitochondrial messenger RNAs form essential subunits of the oxidative phosphorylation complexes I to V except complex II. These complexes play a key role in mitochondrial oxidative phosphorylation, influencing electron transfer and ATP generation [58, 59]. The principal sites for oxidative phosphorylation and ATP generation are mitochondria, mitochondria supply about 95% of the energy needed for cellular functions [60]. Under hypoxic conditions, cells rely solely on glycolysis for ATP production, which leads to the conversion of pyruvate into lactic acid. In aerobic conditions, glycolysis produces pyruvate that enters the tricarboxylic acid (TCA) cycle, also known as the citric acid or Krebs cycle, facilitating mitochondrial ATP synthesis [61].
Mitochondria generate circulating metabolites from the TCA cycle that influence cellular fate and function. TCA cycle metabolites have traditionally been regarded as by-products of cellular metabolism. These metabolites are crucial for the biosynthesis of macromolecules, including nucleotides, proteins and lipids. While fundamental to cellular homeostasis, these processes occur rapidly. It has been recognized that metabolites in the TCA cycle are also involved in controlling chromatin modification, DNA methylation and change of function post-translational protein modification [62].
Mitochondrial morphology exhibits significant variations across different cell types and tissues and swiftly responds to external stimuli and metabolic signals such as nutritional status. The structure of mitochondria, characterized by continuous fission and fusion of their inner and outer membranes, has been intensively studied for its functional implications. Unregulated fission leads to mitochondrial disruption, commonly linked with metabolic dysfunction and disease. Controlled fusion produces stable mitochondrial networks, which counteract metabolic damage, maintain cellular integrity, and inhibit autophagy [63]. Mitochondrial morphology undergoes constant alterations through a dynamic interplay of fusion, fission and cytoskeletal trafficking. The balance between fission and fusion rates determines both the extent to which mitochondria form closed networks and their length. Moreover, these rates are controlled by mitochondrial metabolism, harmful conditions and their cellular environment. Fusion and fission are significant for mitochondrial redistribution, growth, and maintaining a robust mitochondrial network. Moreover, fission and fusion process of mitochondria exert a significant role in disease-associated procedures, including mitochondrial apoptosis and autophagy.
Three members of the Dynamin family play an important role in fission and fusion mechanisms [64]. As dynamic organelles, mitochondria can fuse and divide. These continuous and simultaneous processes are mediated by nuclear DNA-coding proteins, which act upon the mitochondrial membrane. The balance of fission and fusion dictates mitochondrial morphology, adapting to the metabolic demands of the cell. Additionally, these two processes are essential for optimizing mitochondrial bioenergetic and functional capabilities [65]. Maintaining a healthy network of interconnected mitochondria is crucial for ensuring the proper positioning of mitochondria towards high-energy-demanding neurons, as well as safeguarding neurons by minimizing oxidative stress. To achieve these objectives, it is imperative to achieve an equilibrium between mitochondrial fusion and fission processes [66].
Implications can be observed in conditions like AD and cerebral
ischemia/reperfusion, where microglial mtDNA undergoes mutations or deletions
[67, 68]. Recent research has reported a correlation between levels of microglial
mtDNA deletions in distinct brain regions with varying pathological
susceptibilities to AD and their corresponding disease stages
[69].
Once taken up
by nearby microglia, mtDNA will binds to cyclic guanosine 5
Accumulating in vitro and in vivo evidence suggests that
metabolic impairments in the CNS contribute to both microglial activation and
cell death, exacerbating pathology in neurodegenerative diseases [72]. Oxidative
stress and reactive oxygen species generation are pivotal mechanisms in this
process [72, 73]. Microglia are attacked by oxidative stress, and reactive oxygen
species, leading to mitochondrial dysfunction. Microglia with mitochondrial
dysfunction increase mitochondrial damage, increase the release of inflammatory
factors into the extracellular space and overproduce ROS. These pathological
changes further increase the inflammatory response of microglia. Considerable
proteomic analyses of AD brains and cerebrospinal fluid have demonstrated
elevated levels of proteins linked to glucose metabolism, coinciding with
early-stage microglial activation [74]. Upon inflammatory stimulation, microglia
shift their metabolic pathway to less efficient glycolysis, potentially
undermining their metabolic efficiency and phagocytic capability, ultimately
leading to microglial dysfunction and A
An increasing body of literature highlights the significant role of mitochondrial fusion/fission imbalance in microglia-mediated neuroinflammation in AD. Evidence has demonstrated that activated microglia in AD mouse models exhibit mitochondrial damage due to dysregulated mitochondrial dynamics [78]. In neurodegenerative conditions, neuronal debris is believed to initiate glial-mediated neuroinflammation, contributing to further neuronal loss. Notably, this phenomenon has been specifically observed in microglia. The ratio of damaged to functional mitochondria released from microglia and the resultant neuronal damage, are both regulated by Fis1-mediated mitochondrial fragmentation within these glial cells [79].
Recent studies have suggested that defective mitophagy might play a role in the development of AD. Further research has shown that mitophagy is impaired in the hippocampi of patients with AD, as well as in induced pluripotent stem cell-derived human neurons and animal models of the disease [80].
Mitochondrial dysfunction can trigger diverse signaling pathways, leading to the metabolic reprogramming of glial cells and exacerbate pathogenic events related to AD [81]. Signaling pathways associated with mitochondrial dysfunction in microglia are showed in Fig. 1 and Table 1.
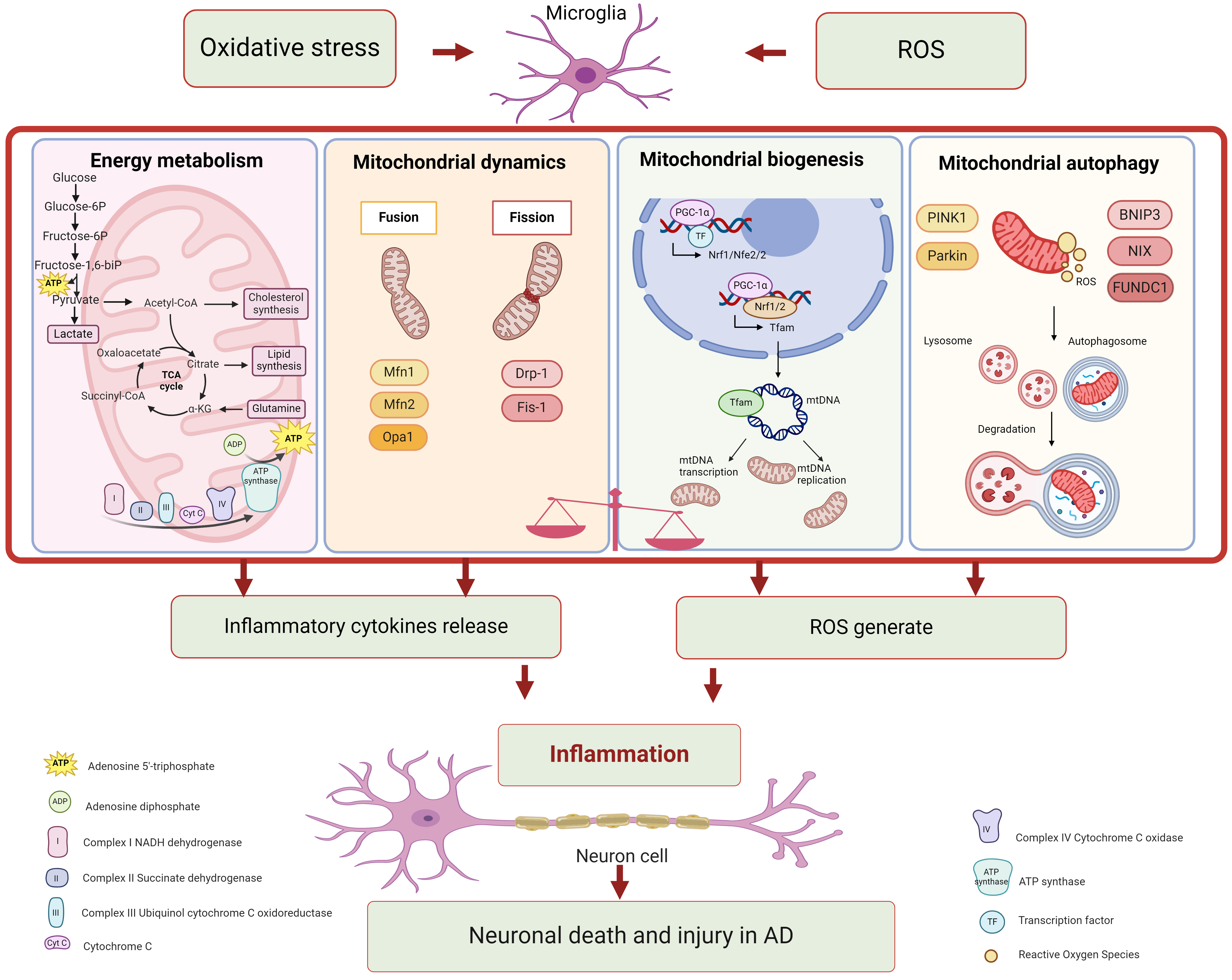 Fig. 1.
Fig. 1.Mitochondrial dysfunction in microglia associated with
Alzheimer’s disease. Mitochondria perform multiple roles, including energy
metabolism, mitochondrial dynamics, mitochondrial biogenesis, and mitochondrial
autophagy. Each function participates in various signaling pathways. Microglia
with mitochondrial dysfunction significantly contribute to neuroinflammation in
Alzheimer’s disease. Figure was made by biorender
(https://www.biorender.com/).
ROS, reactive oxygen species; ATP, denosine triphosphate; Acetyl-CoA, acetyl
coenzyme A; TCA, tricarboxylic acid cycle;
| Biological process | Physiological environment | Physiological environment |
| OXPHOS | Main way of generating energy | Complex I, III, IV activity decreased; ROS increased |
| Glycolysis | It produces almost no energy | Fast energy supply in response to inflammation or stress |
| TCA cycle | Normal | PDH, 2OGDH, glutamate, glutamine, GABA, and NAA decreased |
| Mitochondrial fusion | Normal | Tomm40, Opa1, Mfn1, Mfn2 decreased |
| Mitochondrial fission | Normal | Drp1, Fis1 increased |
| PGC-1 |
Normal | TFAM, Nrf1, Nrf2, and PGC-1 |
| Mitophagy | Normal | PINK, Pakin, LC3 decreased |
TCA, Trica rboxylic acid; OXPHOS, oxidative phosphorylation; ROS, reactive
oxygen species; PDH, Pyruvate dehydrogenase; 2OGDH, 2-oxoglutarate dehydrogenase;
GABA, Gamma-aminobutyric acid; NAA, N-acetyl-aspartate; Tomm40, Translocase of
outer mitochondrial membrane 40; Opa1, Optic atrophy 1; Mfn1, mitofusin-1; Mfn2,
mitofusin-2; Drp1, dynamin related protein 1; Fis1, mitochondrial fission 1
protein; TFAM, mitochondrial transcription factor A; Nrf1, Nuclear respiratory
factor 1; Nrf2, Nuclear respiratory factor 2; PGC-1
Mitochondrial energy metabolism encompasses multiple processes, including oxidative phosphorylation, the TCA cycle and glycolysis. Mitochondria serve as the primary site for oxidative phosphorylation, where the electron transport chain synthesizes ATP. This chain is mainly composed of Complexes I to V, located in the inner mitochondrial membrane [82]. Glycolysis comprises ten reactions that are divided into two phases: ATP investment and ATP payoff. These reactions take place in the cytosol [82]. The mitochondrial matrix is responsible for the production of the TCA cycle, which consists of eight enzymatic steps that consume and regenerate citrate [62]. The researchers observed a decline in mitochondrial complex I, III, and IV activity in AD models or the brains of AD patients. Furthermore, levels of TCA cyclically derived metabolites (glutamate, Gamma-aminobutyric acid (GABA), N-acetyl-aspartate (NAA), and glutamine) were found to be significantly reduced in the AD model, which correlates with the pathology of AD [83]. Dysfunction in the energy metabolism of mitochondria and metabolic reprogramming in microglia have a strong association with AD [84].
Mitochondria are active organelles that exist dynamically in networks. They are constantly brought together and then separated by a process of fusion and fission. A previous study has revealed elevated expression of the mitochondrial fission genes mitochondrial fission 1 protein (Fis1) and dynamin related protein 1 (Drp1), alongside decreased expression of mitochondrial fusion genes Translocase of outer mitochondrial membrane 40 (Tomm40), Optic atrophy 1 (Opa1), mitofusin-1 (Mfn1) and mitofusin-2 (Mfn2) [85]. Research indicates that an imbalance in mitochondrial fusion and fission significantly contributes to microglia-mediated neuroinflammation in AD [84].
Peroxisome proliferator-activated receptor gamma coactivator 1
alpha (PGC-1
The signaling pathway that regulates mitophagy is the phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1)/Parkin axis. In depolarized and damaged mitochondria, PINK1 accumulates on the outer membrane of the mitochondria and enlists the E3-ubiquitin ligase Parkin [88, 89]. Parkin contributes to the removal of damaged mitochondria by ubiquitinating proteins in the outer mitochondrial membrane, which include Mfn1, Mfn2, and voltage-dependent anion channel 1 (VDAC1) [82]. Investigation has observed that levels of PINK1, microtubule-associated proteins light chain 3 (LC3), and Lon protease 1 (LONP1) were up-regulated in animal models and the brains of AD patients. Emerging evidence suggests that mitochondrial autophagy, or mitophagy, takes part in the inflammatory responses and phagocytic activities of microglia [80, 90].
In neurodegenerative diseases, mitochondrial dysfunction often precedes the accumulation of toxic proteins, establishing a vicious cycle of deteriorating cellular conditions. Consequently, mitochondria have become prime targets for the development of novel therapeutics in neurodegenerative diseases.
Here, a summary of literature from the past five years is complied into Table 2 (Ref. [91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125]) and Fig. 2, more than 40 candidate molecules were categorized by different drug targets. It shows the treatment of AD by drugs is targeting mitochondrial energy metabolism, mitochondrial homeostasis, mitochondrial biogenesis and mitochondrial autophagy. Encouragingly, some drugs have already entered clinical trials. Rapamycin, targeting mitochondrial energy metabolism, was in phase 2 clinical trials. And metformin, a small molecule targeting mitochondrial biosynthesis, was in phase 3 clinical trials. Perhaps in the near future, these candidate molecules are expected to become effective drugs for the treatment of AD.
| Targets | Drugs | Mechanisms | References |
| Energy metabolism | Vitamin E, Vitamin C, Alpha-lipoic acid, MitoQ, Curcumin, Astaxanthin, Ginkgo biloba preparations | Reduce superoxide radicals and scavenge hydrogen peroxide | [91, 92, 93, 94] |
| Sodium rutin | Enhances the level of mitochondrial oxidative phosphorylation | [95] | |
| Batrachocholine | Improves mitochondrial ATP metabolism and modifying antioxidant proteins | [96] | |
| Ginsenoside Rb1 | Inhibition of mitochondrial complex I activity | [97, 98] | |
| Interferon- |
Boosts glycolytic metabolism | [99] | |
| Thy and 2’-De | Increase ATP production and inhibited glycolysis | [100] | |
| Naringin | Reduces oxidative damage | [101] | |
| Rapamycin, Torin1 and Niclosamide | Downregulation of the mTORC1 pathway | [102] | |
| Myricetin (Myr) | Inhibits P38 MAPK pathway activation | [103] | |
| BPTES | Glutaminase inhibitor | [104] | |
| Mesenchymal stem/stromal cells (MSCs) | Secret neuroprotective and anti-inflammatory factors to promote the survival of neurons and transfer functional mitochondria and miRNAs to boost their bioenergetic profile as well as improve microglial clearance of accumulated protein aggregates | [105] | |
| GRP75 | Improves mitochondrial function and decrease oxidative stress | [106] | |
| Mitochondrial dynamics | Mdivi-1 | Repairs mitochondrial breakage and distribution defects | [107] |
| Butylphthalide | Balances the mitochondrial dynamics | [108] | |
| AIII | An inhibitor of JAK2 | [109] | |
| Cannabidiol | Increases the expression of Mfn2 | [110] | |
| Mitochondrial biogenesis | Resveratrol | Sirtuin proteins activator | [111] |
| Quercetin | Promotes PGC-1 |
[112] | |
| Ginsenoside RC | Activates the SIRT1-pgC1 |
[113] | |
| Berberine | AMPK activator | [114] | |
| Metformin | Enhances the transcriptional activity of mitochondrial biogenesis factor Nrf-2 and thus upregulating the expression of peroxidase GPx7 | [115] | |
| Mitochondrial autophagy | Melatonin | Improves mitochondrial-lysosomal fusion | [116] |
| Increases the expression of PINK1, Parkin and Beclin-1 | [117] | ||
| Lithium urate, Tomatidine, Rhapontigenin, Urolithin A, Actinonin, Nicotinamide riboside, T-271 | Mitophagy enhancers | [118, 119, 120] | |
| Ac-YVAD-cmk | Caspase-1 inhibitor | [121] | |
| VBIT-4 | VDAC1 inhibitor | [122] | |
| VRP, Nicardipine | Ca |
[123, 124] | |
| Quercetin | Inhibition of mtROS-mediated NLRP3 inflammasome activation in microglia through promoting mitophagy | [125] |
ATP, denosine triphosphate; mitoQ, mitoquinone; mTORC1, mammalian target of
rapamycin complex 1; MAPK, mitogen-activated protein kinase; BPTES,
bis-2-(5-phenylacetamido-1, 3, 4-thiadiazol-2-yl) ethyl sulfide; GRP75,
glucose-regulated protein 75; Mdivi-1, Mitochondrial division inhibitor-1; JAK2,
Janus kinase-2; Mfn2, mitofusin 2; PGC-1
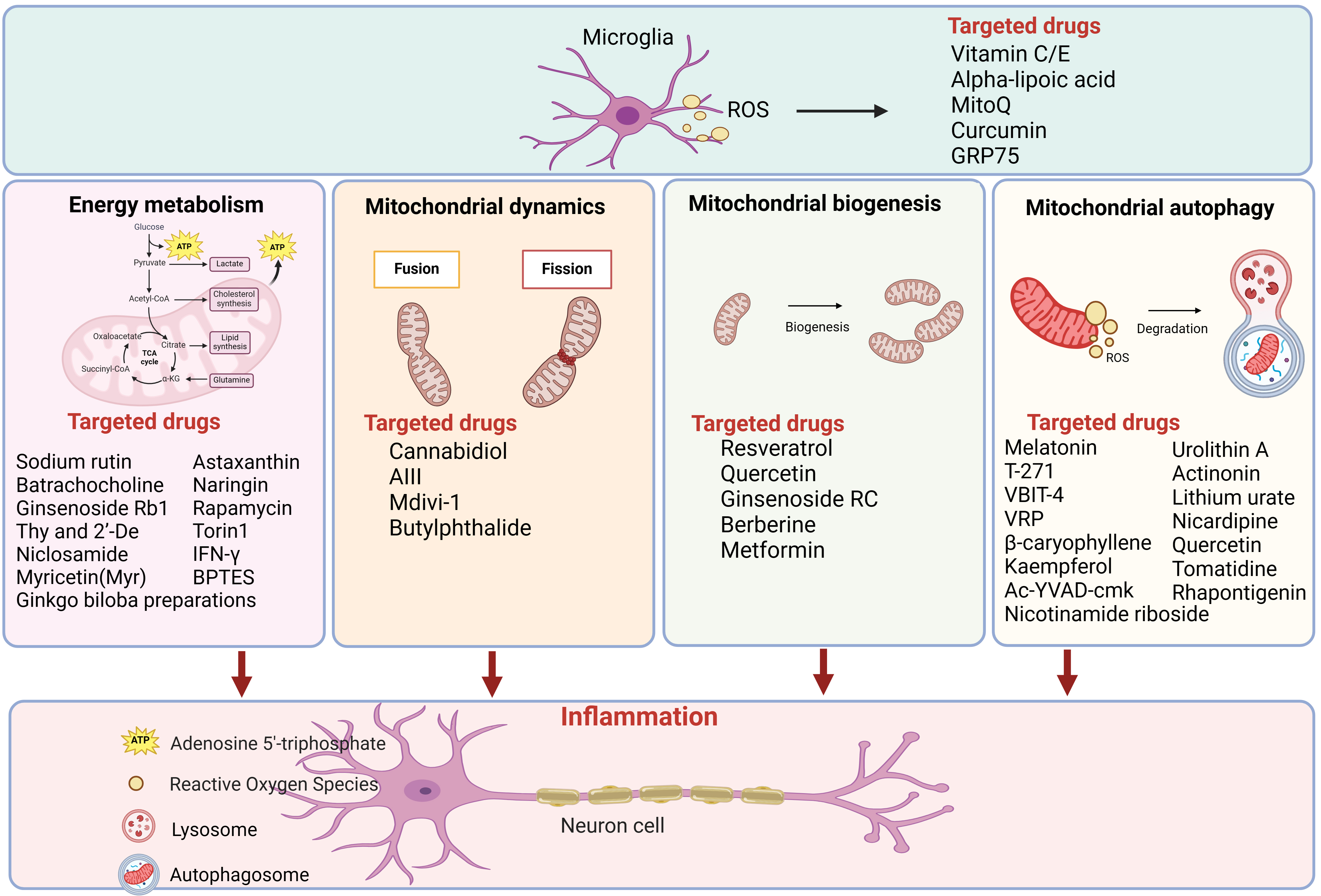 Fig. 2.
Fig. 2.Drugs categorized by mitochondrial energy metabolism,
mitochondrial biogenesis and mitochondrial autophagy for AD therapy. More than
40 candidate molecules were showed in the figure, which have demonstrated
promising results in preclinical AD models. Figure was made by biorender
(https://www.biorender.com/). ROS, reactive
oxygen species; GRP75, glucose-regulated protein 75; ATP, denosine triphosphate;
acetyl-CoA, acetyl coenzyme A; IFN-
Recent studies have demonstrated promising results in preclinical AD models. Some of these drugs have also been used in healthy or AD patients at various stages of clinical trials. However, there are many challenges between preclinical success and clinical application. Initially, it must be determined whether drugs are safe and whether they cause serious adverse reactions. Subsequently, do all drug candidates cross the blood-brain barrier? If not, can nanotechnology be employed as a vehicle to help the drug translocate? Further, can these drugs target the mitochondria that reach microglia and are there an off-target effects? Is a guide-RNA required for localization? Can the bioavailability of these drugs following entry through the blood-brain barrier reach an effective concentration? Finally, do these drugs have an effect on nerve cells other than microglia? These issues still need to be addressed. Further study of the mechanisms of microglia mitochondrial dysfunction in AD is required to provide ideas and directions for such clinical research.
In this article, the role of mitochondrial dysfunction in microglia in AD is reviewed and the preclinical drugs targeting mitochondria for AD in the past five years are summarized. It is proposed that mitochondria may be a potential target for AD treatment and the preclinical drugs targeting mitochondria for AD in the past five years are summarized. However, there is still a long path from preclinical research to clinical application and further research on mechanisms of mitochondrial dysfunction in microglia are needed.
XZ, and FZ and YX formulated the original idea and modified the manuscript. YL and TL organized documents, wrote the manuscript and designed and draw the figures and tables. CL, TC and WY prepared the tables and figures and helped with paper revision. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by research grants from Shenzhen Science and Technology Program (JCYJ20210324111213037) and China Postdoctoral Science Foundation (2023M733658).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.